Bardet -Biedl Syndrome, also known as Laurence-Moon-Biedl-Bardet Syndrome (LMBBS), is a ciliopathy disease that is exclusively hereditary. The syndrome manifests itself in the form of multiple malformations that are triggered by changes (mutations) in different gene locations or chromosomes.
What is Bardet-Biedl Syndrome?
In adiposity (obesity), the body exhibits a pathological accumulation of fatty tissue. In the case of BBS, pathologically increased accumulations of fat on the legs, abdomen, buttocks, arms, chest and hips occur predominantly as trunk obesity, with the torso, legs and thighs being particularly badly affected. See AbbreviationFinder for abbreviations related to LMBBS.
The clinical picture defined by the physicians Moon and Laurence and later by Bardet and Biedl is a disease in which retinal dystrophy occurs as a medically significant feature in combination with other symptoms. Due to this complicated initial medical situation, the conclusive determination of BBS disease is difficult. In 1866, this clinical picture was first recorded medically.
Four people studied had retinitis pigmentosa (retinal dystrophy, RP) associated with paraplegia (spastic paralysis) as well as hypogenitalism (underdeveloped sex organs) and intellectual disability. In 1920, the French physician Bardet described a disease consisting of RP (retinal dystrophy), hypogenitalism, polydactyly and obesity.
The Prague pathologist Biedl also found debility (mental confusion). In 1925, the researchers Weiss and Solis-Cohen summarized the known cases and called the clinical picture Laurence-Moon-Biedl-Bardet syndrome.
Causes
In the following years, the medical literature increasingly pointed out that the cases recorded by Laurence and Moon are a rare special form that only occurs in isolated cases together with BBS. Recent medical research results assign the Bardet-Biedl syndrome to the field of ciliopathies (cilia diseases).
These diseases record a common dysfunction of the so-called cilia (small projections, antennae) that appear on the majority of cells in the human organism. The ciliopathies are characterized by smooth transitions and overlaps between different cilia diseases.
Symptoms, Ailments & Signs
The main feature of hereditary retinal dystrophy is a generic term that describes the onset of loss of function and subsequent degeneration (destruction) of the photoreceptors. They lead to a progressive (progressive) loss of visual function. The rapidly progressive visual disturbances usually appear very early in children, when they are between four and ten years old. They manifest themselves in different ways, depending on the photoreceptors affected.
As a “rod-cone form” with the characteristic course of retinitis pigmentosa (RP), the disease has its origins in the retinal periphery (outer retina) and develops through progressive visual field loss into macular degeneration (destruction of sharp vision).
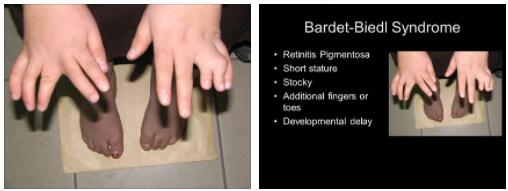
In adiposity (obesity), the body exhibits a pathological accumulation of fatty tissue. In the case of BBS, pathologically increased accumulations of fat on the legs, abdomen, buttocks, arms, chest and hips occur predominantly as trunk obesity, with the torso, legs and thighs being particularly badly affected. Polydactyly is a prominent symptom and a significant feature of Bardet-Biedl syndrome. The finding is not easy, since the rudimentary polydactyly is surgically corrected after birth.
X-rays are able to provide further information. Polydactyly can present with different signs, such as rudimentary toe or finger attachment. A toe or finger can be additionally or only partially formed. Unilateral hexadactyly on the foot and/or hand has an additional segment, bilateral hexadactyly occurs on both feet and/or hands.
Toes or fingers that have grown together (syndactyly) and shortening of one or more toes or fingers (brachydactyly) are also signs of BBS. Only a few patients have all four affected extremities. Mental retardation varies in severity. Only a small number of those affected have a serious intellectual developmental delay. A normally trained intelligence is possible.
Children reach the ability to speak and walk late, and they sometimes show behavioral problems such as anxiety disorders. Compulsive or autistic behaviors, a low frustration threshold and unstable emotionality are other possible side effects. What is known is preferred, while changes are rejected. Abnormalities in the internal and external sex organs are common.
Other changes include hypospadias (the opening of the urethra is above or below the penis instead of at the front of the penis), abdominal or groin testicles, urethral strictures, foreskin strictures, and posterior urethral valves. In female patients, vaginal atresia (vagina is not patent), missing urethral openings and reduced labia minora are known.
It is not uncommon for the affected women to have irregular menstrual cycles. Kidney changes are common side effects. The finding depends on the examination of the urinary tract and the kidneys using ultrasound (sonography).
Diagnosis & course of disease
Bardet-Biedl Syndrome (BBS) has six main symptoms, which do not always appear together. The doctors assume a corresponding finding if at least four of the main symptoms are present. Alternatively, there is a high probability of the disease being present if the patient shows three main symptoms and two secondary symptoms.
The six main symptoms are retinal dystrophy, obesity (abnormal accumulation of fatty tissue, overweight), polydactyly (excess toes and/or fingers), mental retardation (mental delay), hypogenitalism (underdeveloped sex organs) and kidney disease. Secondary symptoms that occur with low frequency include speech delays, speech deficits, cardiac malformations, ataxia (impaired movement coordination), asthma, diabetes mellitus (diabetes), Crohn’s disease (inflammation of the large and/or small intestine), rib and vertebral dysplasia and kyphoscoliosis (spinal curvature). on.
Complications
Those affected usually suffer from a loss of visual function as a result of Laurence-Moon-Biedl-Bardet syndrome. The loss does not occur suddenly, but gradually. In the worst case, those affected suffer complete blindness, which can usually no longer be treated.
Especially in young people and children, blindness can lead to severe mental health problems or even depression. The patients are significantly restricted in their everyday life and suffer from a severely reduced field of vision. In many cases, the Laurence-Moon-Biedl-Bardet syndrome also leads to behavioral problems, so that children in particular can suffer from bullying or teasing.
The development of children is also significantly delayed and limited by the syndrome. Anxiety disorders can also occur. It is not uncommon for the Laurence-Moon-Biedl-Bardet syndrome to cause psychological problems and depression in family members or parents. Unfortunately, causal treatment of the Laurence-Moon-Biedl-Bardet syndrome is not possible.
Some complaints can be limited. However, a completely positive course of the disease does not occur. The patient’s life expectancy is not reduced by the syndrome. In some cases, those affected are sometimes dependent on the help of other people in their everyday life.
When should you go to the doctor?
Since Laurence-Moon-Biedl-Bardet syndrome is a hereditary disease, it can be diagnosed in the womb. At the latest after the birth, a doctor should be consulted if typical symptoms such as blurred vision or obesity are noticed. Malformations of toes and fingers are also a clear indicator of a disease. Parents who notice such symptoms in their child should inform the pediatrician immediately.
A comprehensive examination provides information about the disease. The therapy is then usually initiated directly, which consists of various treatments by orthopaedists, neurologists, ophthalmologists, internists and therapists as well as physiotherapistscomposed. Further visits to the doctor are necessary if the treatment does not have the desired effect. Medical advice is also required in emergency situations, such as when the child falls as a result of a deformity or suddenly suffers from a seizure. If the patient shows signs of psychological problems, the parents must consult a suitable therapist. Older children can contact the school psychologist together with their parents and discuss appropriate measures.
Treatment & Therapy
This disease occurs on the basis of autosomal recessive inheritance, which means that both copies (alleles) of a BBS gene have a change (mutation). The patient’s parents are “hybrid” and each carries an altered and an unchanged allele of the corresponding gene. They don’t have the disease. Children only become ill if father and mother pass on the mutated allele. In other children, the probability of recurrence is 25 percent.
A causal therapy option is not yet known, since certain symptoms of the disease cannot yet be definitively assigned to the different gene mutations. The symptoms and their manifestations appear differently even in sick siblings. Since the characteristic full picture of BBS is only present in rare cases, especially in infancy, a corresponding finding is difficult.
Due to the frequently present oligosymptoms, with which very few atypical and only mildly pronounced symptoms occur, other possible clinical pictures must be considered in the differential diagnosis. Changes in the same gene can lead to various clinical pictures, such as Joubert, Bardet-Biedl or Meckel-Gruber syndrome.
Outlook & Forecast
The overall prognosis in the presence of Laurence-Moon-Biedl-Bardet syndrome is rather poor because the multiple malformations are congenital and incurable. If four of the six key symptoms occur, the diagnosis of Laurence-Moon-Biedl-Bardet syndrome is secured. In addition to the main symptoms, there are numerous secondary symptoms. This includes a gradual onset of blindness.
Due to the complexity of the symptoms, there is no prospect of healing. There is only a mediocre chance of noticeable relief from symptoms. The number of possible malformations and disorders in Laurence-Moon-Biedl-Bardet syndrome is so large that the hereditary disease is difficult to treat. The course of this genetic disease cannot be influenced anyway. However, some of the presenting symptoms can be alleviated.
However, the overall poor prognosis does not reduce the life expectancy of the people affected. At an advanced age and after blindness, those affected may be permanently dependent on help or care. Through interdisciplinary medical efforts, many sufferers of Laurence-Moon-Biedl-Bardet syndrome can experience a somewhat milder course of the disease.
The increasing visual problems represent a problematic part of the disease that is difficult to treat. Increasing visual impairments already occur in the small children affected. They deteriorate over time. However, the vision problems do not necessarily lead to blindness in all those affected. The psychological consequences of Laurence-Moon-Biedl-Bardet syndrome can usually be treated well.
Prevention
Prevention in the sense of preventing this disease is not possible. Regular monitoring of the symptoms and side effects that occur is important. Repeated checks of blood pressure and kidney function, nutritional advice, physiotherapy and ergotherapy as well as speech therapy are possible therapeutic approaches.
Aftercare
In most cases, those affected by Laurence-Moon-Biedl-Bardet syndrome have no special options for aftercare, so that a doctor should be contacted and consulted very early on with this disease. As a rule, self-healing cannot occur, so that treatment by a doctor is always necessary.
Since the Laurence-Moon-Biedl-Bardet syndrome is a hereditary disease, the person concerned should have a genetic examination and counseling carried out if they wish to have children, so that the Laurence-Moon-Biedl-Bardet syndrome does not pass on to their offspring is passed on. In many cases, those affected are dependent on surgical interventions in order to alleviate the malformations and deformities.
The person concerned should definitely rest after the procedure and take care of their body. In any case, exertion or other physical and stressful activities should be avoided in order not to unnecessarily burden the body. Since Laurence-Moon-Biedl-Bardet syndrome can also lead to abnormal behavior, parents should support and encourage the child’s development. Loving and intensive conversations with the child are also necessary in order to prevent psychological upsets or depression.
You can do that yourself
Laurence-Moon-Biedl-Bardet syndrome has various symptoms, with patients often suffering the most from visual impairment. Even in children, the usual ability to see begins to deteriorate, so it is the parents who present the child to a doctor and thus speed up the diagnosis. In this way, the disease can be treated quickly, although the treatment options have so far only been of a symptomatic nature.
The visual disturbances become increasingly worse in the sick children and thus significantly impair everyday life, so that the quality of life of those affected decreases. Because of the vision problems, numerous difficulties develop for the patient when attending school, in their free time and with regard to their physical integrity. The risk of accidents also increases significantly, for example on the road. That is why parents accompany their sick children whenever possible or hire nursing staff so that the patient is not left to their own devices.
In some cases, the disease progresses to blindness. Since such a development is already apparent beforehand, the patients are preparing for it. The parents rearrange the living space so that it does not contain any sources of danger for the visually impaired person. In addition, the blind people affected learn how to use a cane so that they can move about independently outside their own home.